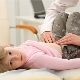
Spinální svalová atrofie u dětí

Spinální svalová atrofie je závažná patologie, která často dělá jednoduché akce, jako je chůze, sezení, nepřístupné pro dítě. Dítě může být zbaveno i takové přirozené příležitosti jako nezávislé dýchání. Je velmi obtížné předpovědět pro tuto patologii, koneckonců neexistuje ani speciální léčba ani profylaxe, ale vše závisí na formě onemocnění a faktorech, který lék nemůže najít vysvětlení.
Co je to?
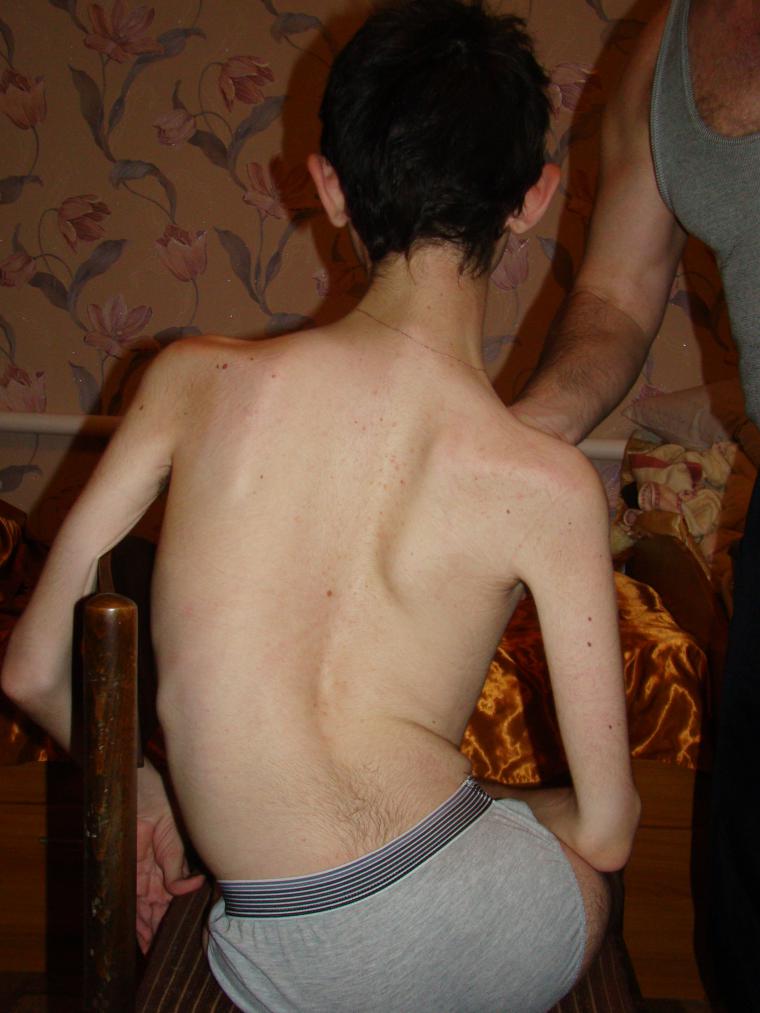
Když už mluvíme o spinální svalové atrofii, neznamenají ani jednu specifickou chorobu, ale celou skupinu onemocnění pod všeobecnou zkratkou CMA. Všechny jsou dědičné a jsou spojeny s degenerací nervových buněk míchy, které jsou zodpovědné za motorické funkce.
Mezi genetické patologie u dětí spinální svalové atrofie zaujímají přední místa z hlediska četnosti šíření. A asi jeden z 6 tisíc dětí se narodí s takovou hroznou diagnózou. V 50% případů děti nežijí do věku dvou let a umírají. Zbytek života je postižení.

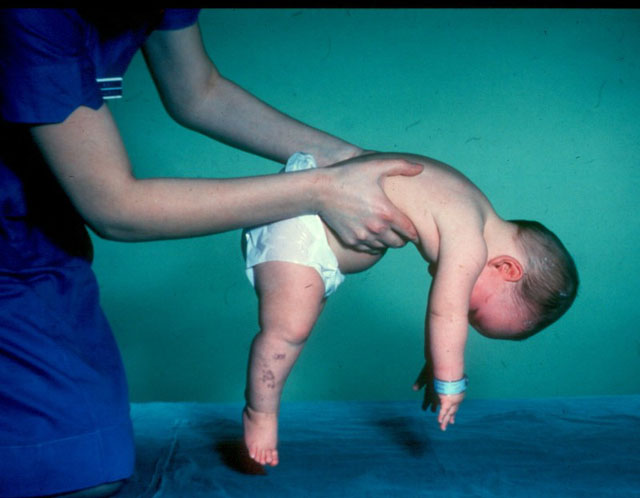
Problém, podle genetiků, je mnohem širší, než by se mohlo zdát z dané statistiky.
Onemocnění se vyvíjí v důsledku mutace určitých genů a jedním z nich je SMN1, který je považován za hlavního "viníka" patologie, v modifikované formě recesivně přítomného v každém padesátém obyvateli planety. To znamená, že zdraví rodiče, kteří si ani neuvědomují, že jsou recesivními nosiči mutovaného genu, mohou mít i dítě s spinální svalovou atrofií.
Skupina nemocí byla poprvé popsána v 19. století Guido Verdingem, jehož jméno bylo později pojmenováno jako jedna z dětských odrůd AGR.
Klasifikace
Nejběžnější forma SMA u dětí je proximální. Jedná se o několik typů onemocnění, z nichž ne všechny se projeví bezprostředně po narození dítěte.
- Verding-Hoffmanova choroba - typ 1 MCA, závažné dětské onemocněníse projevuje v prvních šesti měsících života dítěte. Prognózy s ní nejnepříznivější, většina pacientů zemře. Dítě s SMA typu 1 nemůže stát, ani sedět, ani se převrátit. Mnoho novorozenců má zhoršené reflexy sání a polykání. Často není možnost spontánního dýchání nebo dýchání je obtížné.
- Atrofie Dubovitsa - CMA typ 2, pozdní dítě. To obvykle nastane mezi věky šest měsíců a rok a půl a pozdnější. Dítě nemůže chodit, stát, ale je schopno sedět, jídlo není narušeno, zvládá úkol polykání, sání. Jak dlouho bude dítě žít, závisí na stavu dýchacích svalů.
- Atrofie typů Kugelberg-Welander - CMA 3, infantilní. Obvykle se nachází ve věku jednoho a půl roku, obvykle za dva roky. Prognosticky příznivější forma. Malí pacienti mohou stát, sedět, pohybovat se, ale zažívají velkou slabost, a proto ve většině případů potřebují invalidní vozík, bez něhož je jejich normální životní aktivita pro ně obtížná.
- Kennedyho atrofie - CMA typ 4, bulbospinalnaya. Obvykle považován za dospělou formu, ale příležitostně u dětí po 15 letech. Vliv života je zřídkakdy ovlivňován, oslabování svalů nastává pomalu, postupně, člověk, který vedl normální život a považoval se za zdravého, nakonec se stává invalidním a ztrácí schopnost pohybu nezávisle.


Více či méně známá atrofie Duchenne z první ruky a atrofie Vulpianu jsou „dospělí“ SMA, první je obvykle detekován po 18 letech a druhý po 20 letech.
U dětí se zaznamenávají nejen izolované formy SMA, když se nic netýká kromě svalové dystrofie, ale také kombinovaných forem, kdy spinální atrofie není jedinou diagnózou a dítě má jiné genetické nebo vrozené problémy, jako jsou srdeční a cévní defekty, oligofrenie.
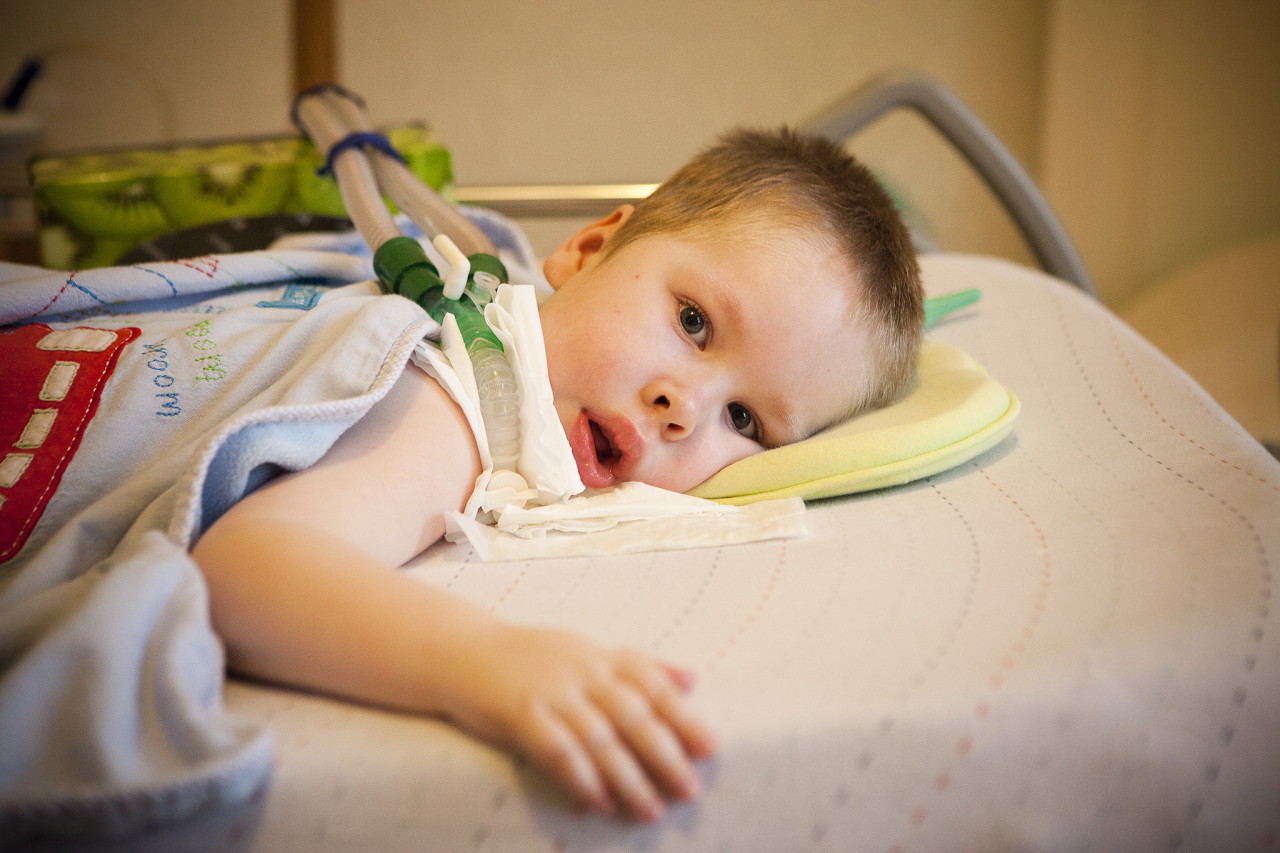
Důvody
Jak již bylo zmíněno, hovoříme o genetickém onemocnění, a proto je důvodem jeho výskytu oblast vyhledávání genetiků. Dítě dědí jeden z recesivních genů na pátém chromozomu (tyto mohou být SMN, NAIP, H4F5, BTF2p44 geny).
Pravděpodobnost přenosu takového genu na potomstvo z nosiče je vysoká - 25%. Pokud jsou maminka i táta skrytými nosiči mutovaného genu, pak je pravděpodobnost AGR u dítěte 50%. Zasažený abnormální gen zabraňuje normální produkci SMN proteinu a nervové buňky zodpovědné za motorické funkce svalů v míše začínají postupně umírat. Proces jejich smrti pokračuje i po narození dítěte.


Projevy
Symptomy závisí na typu nemoci. Protože považujeme pouze čtyři typy dětí, je třeba poznamenat, že svalová slabost a svalová atrofie jsou charakteristické pro všechny. Jinak má každý typ svůj vlastní klinický obraz a charakteristické rysy.
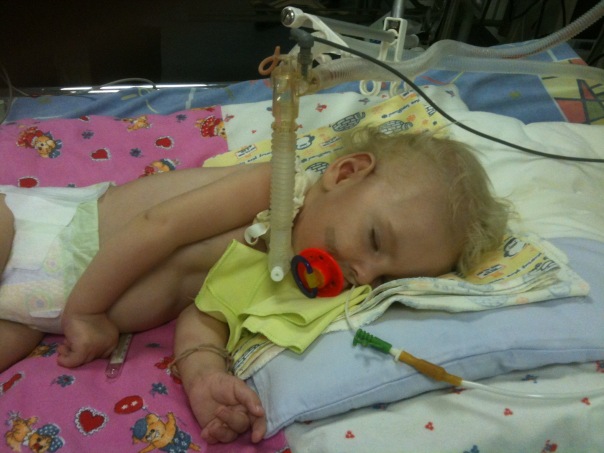
- CMA typ 1 (Verding-Hoffmanova atrofie) k dispozici pro detekci i během těhotenství. Lékař může mít podezření na onemocnění plodu s velmi pomalým mícháním. Potvrzení diagnózy ve stadiu přenášení dítěte je však obtížné, obvykle se to děje po porodu. Dítě s takovou atrofií nemůže držet hlavu sám, házet ze strany na stranu, nesedí. Téměř vždy leží na zádech, jeho pozice je uvolněná, nezvedá nohy, nepřivede je k sobě, nedává si dlaně dohromady. Ve velmi raném stádiu mohou být velké problémy, aby se dítě živilo, protože spolkne, že to dopadá velmi špatně nebo nefunguje. Většina dětí zemře do dvou let. Některým se podaří žít sedm až osm let, ale atrofie se jen zhoršuje. Obvykle k úmrtí dochází v důsledku nedostatečnosti srdce, plic, zažívacích orgánů.
- Typ CMA 2 (atrofie Dubovitsa) při narození se obvykle nedetekuje, protože dítě je schopno dýchat, polykat potravu a teprve po šesti měsících je patrný postup svalové atrofie. Pokud se první příznaky vyskytnou ve věku, kdy se dítě již naučilo postavit se v postýlce, pak je znakem střihu znamení nepřiměřené kapky drobků. Postupně je těžké polykat. Časem dítě začne potřebovat invalidní vozík.
- CMA typ 3 (amyotrofie Kügelberg-Welander) se může objevit v jakémkoli věku po 2 letech věku. Dítě, které se normálně rozrůstalo a rozvíjelo se postupně začíná stěžovat na slabost, obvykle v ramenou, předloktí. Jak postupuje, je pro něj obtížné běžet, chodit po schodech, dřepnout. Vše záleží na péči - někteří si zachovávají schopnost samostatně se pohybovat po mnoho let.
- CMA typ 4 (Kennedyho atrofie) vyskytuje se pouze u mužských pacientů, protože je považován za spojený s pohlavním chromozomem X. První příznaky jsou slabost v oblasti stehenních svalů a postupně jsou ovlivňovány kraniální nervy. Onemocnění postupuje pomalu.

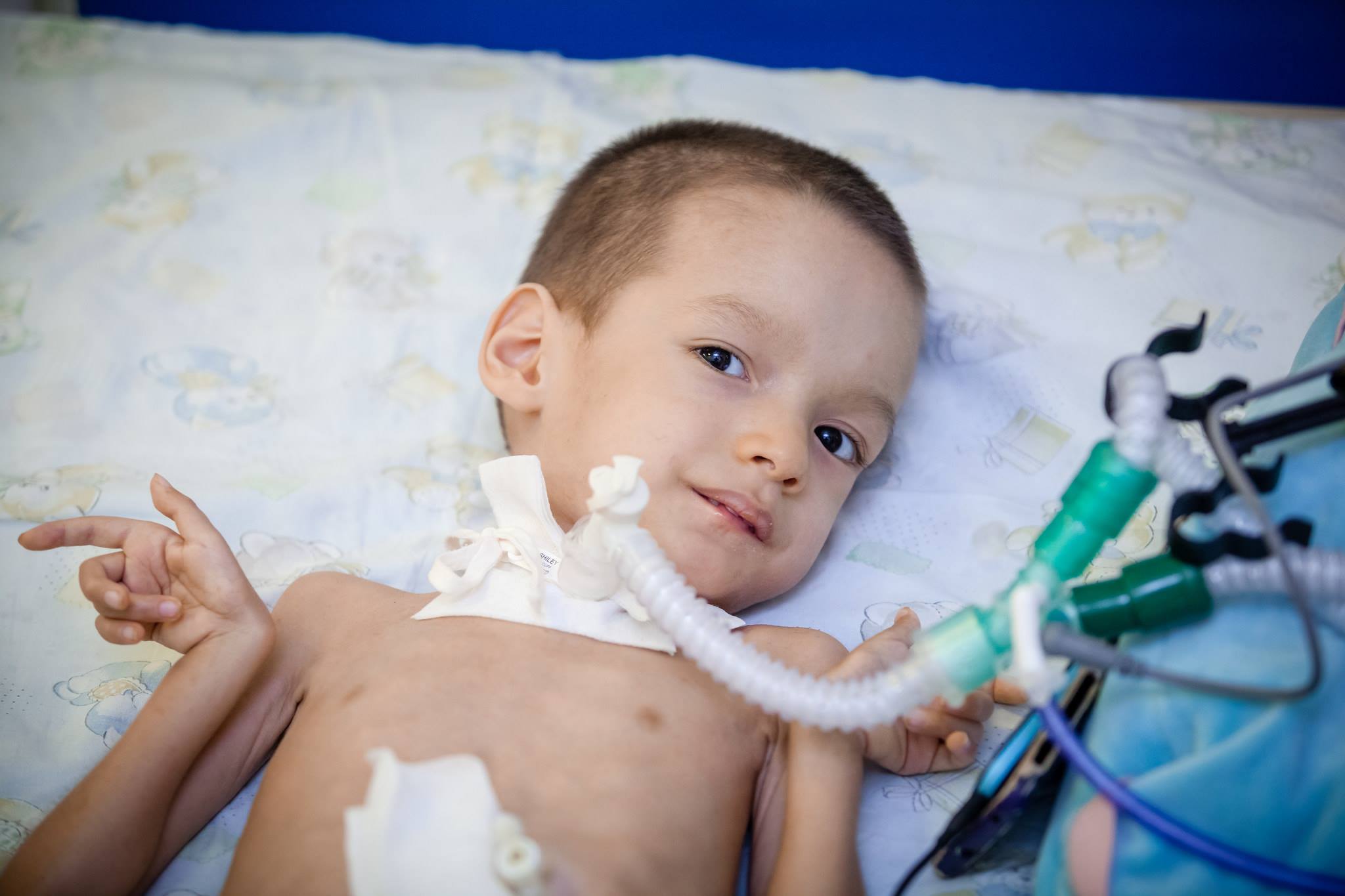
Léčba
Bohužel dnes lékařská věda nemůže nabídnout metody a prostředky pro léčbu SMA. Takové metody neexistují. Aby se zachovaly funkce těla a maximalizovala se doba, po kterou se dítě může pohybovat, léky, jako je Prozerin, Oksazil. Snižují aktivitu enzymu schopného štěpit acetylcholin, který přenáší excitační puls vlákny nervového systému.
Doporučuje se také systematické užívání léčiv, které zvyšují energetický metabolismus na buněčné úrovni, vitamínů skupiny B, nootropních léčiv, jakož i přípravků kyseliny draselné a nikotinové.
Dítě s SMA dieta ukázalaAle nedávné studie ukázaly, že role stravy je poněkud přehnaná - neexistuje žádný důkaz, že vysoký obsah bílkovin v potravinách alespoň nějak ovlivňuje rychlost progrese onemocnění.
Ale s kalorií by měly být opatrnější - vzhledem ke snížené svalové činnosti, může dítě rychle získat další libry.
Pomůže to prodloužit dobu více či méně naplňujícího života léčebná masáž, UHF, elektroforéza, dechová cvičení pro udržování dýchacích svalů, plavání. Doporučujeme nosit podpůrné páteřní nebo hrudní ortopedické přístroje.
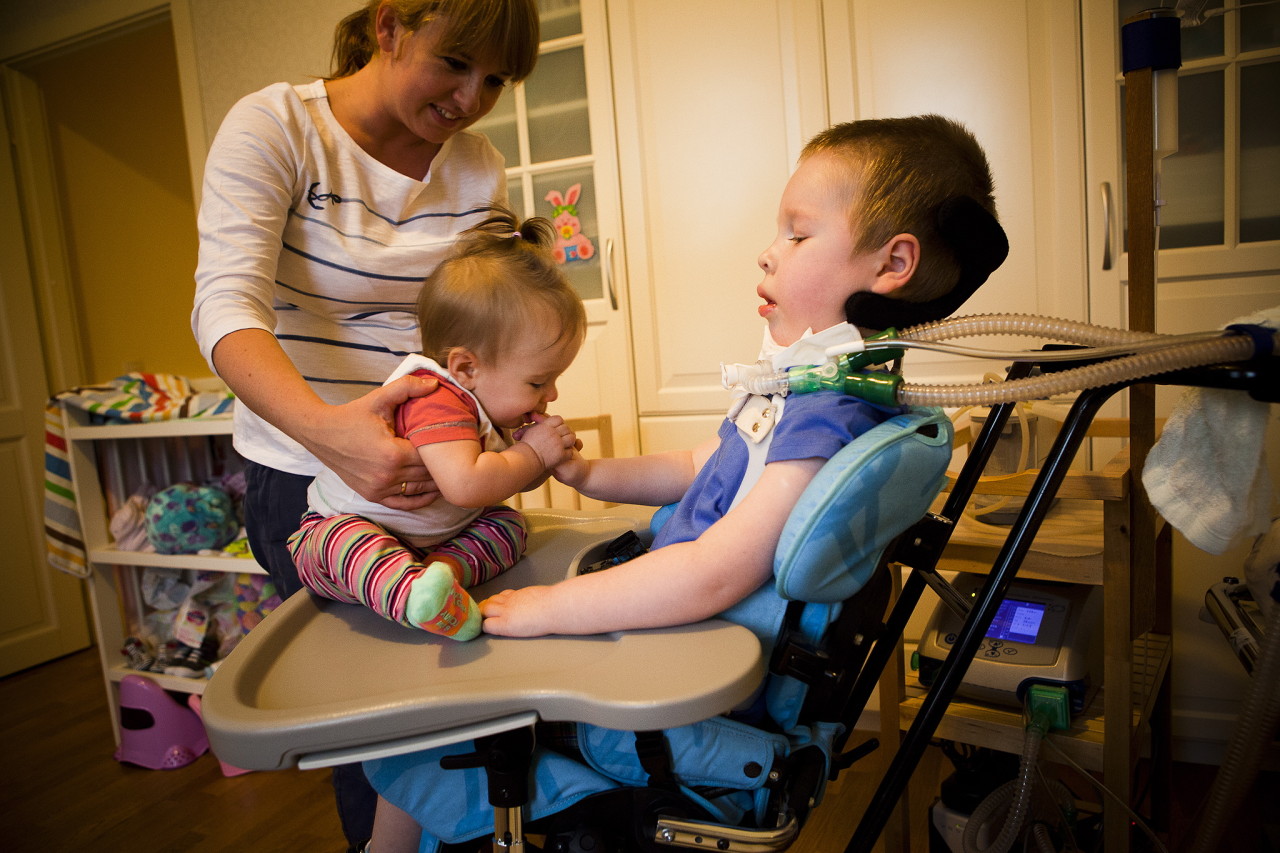
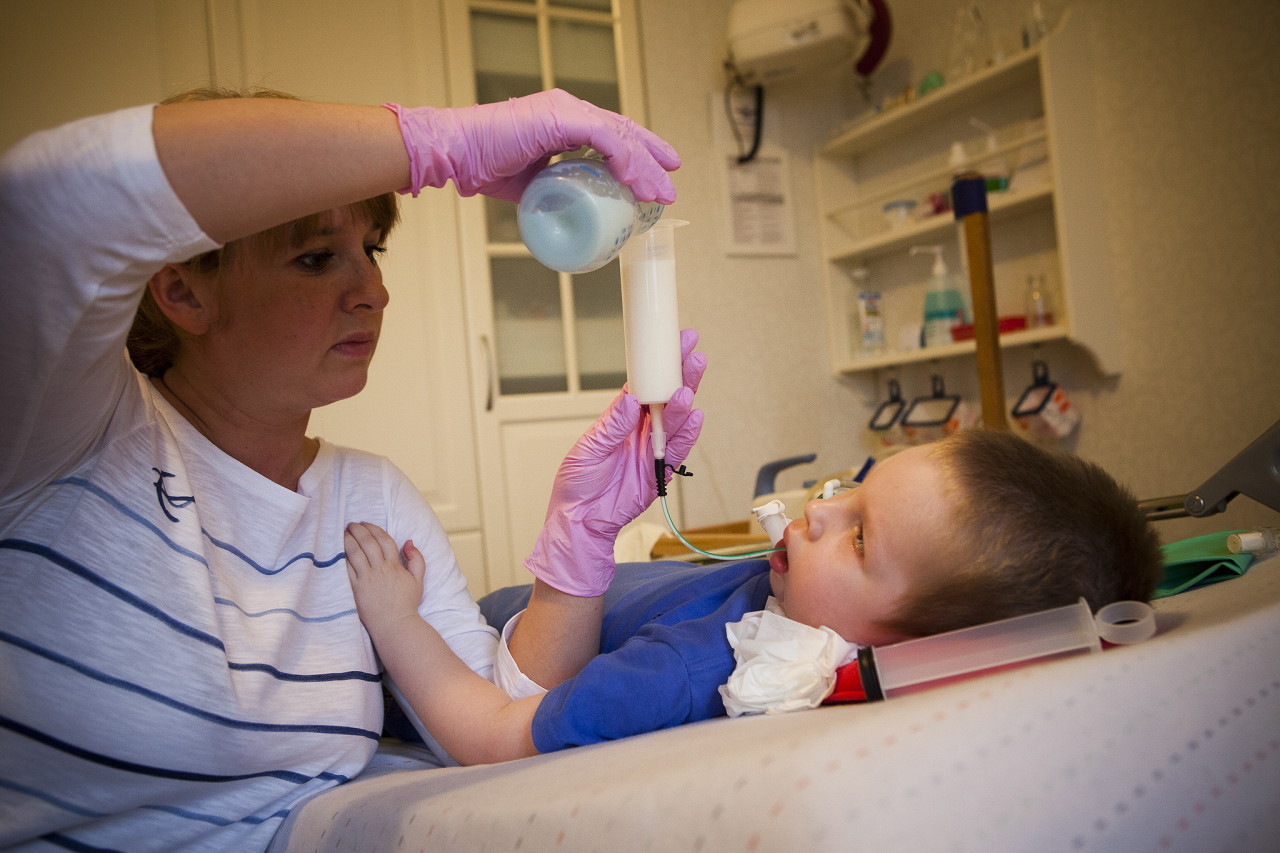

Více informací o této nemoci říká odborník ve videu níže.